
Protein structure introduction - PowerPoint PPT Presentation
1 / 24
Title:
Protein structure introduction
Description:
FSSP, SCOP and CATH databases were screened for all dissimilar domains that ... FSSP assignments compared against CATH, and against SCOP. ... – PowerPoint PPT presentation
Number of Views:271
Avg rating:3.0/5.0
Title: Protein structure introduction
1
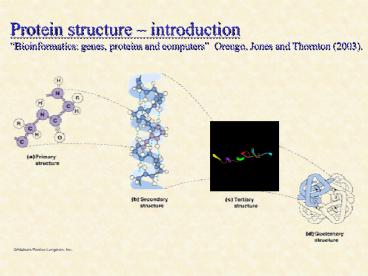
Protein structure introduction Bioinformatics
genes, proteins and computers Orengo, Jones and
Thornton (2003).
2
Secondary structure elements
3
Tertiary structure protein fold
complete 3-dimensional structure
why is it interesting? isnt the sequence enough?
- the structure is more conserved!
- detection of distant evolutionary
- relationships
- a key to understand protein
- function
- Structure-based drug design
4
Fold classification
classification clustering proteins into
structural families
motivation?
- profound analysis of evolutionary mechanisms
- constraints on secondary structure packing?
- classification at domain level
5
CATH Protein Structure Classification
- hierarchical classification of protein domain
structures in the Brookhaven Protein Databank
(PDB). - domains are clustered at four major levels
- Class
- Architecture
- Topology
- Homologous superfamily
- Sequence family
6
CATH hierarchical classification
- Classsecondary structure content mainly
a,mainly b,a b, low 2nd structure content. - Architecturegross orientation of secondary
structures, independent of connectivity. - Topology ( fold)clusters structures according
to their topological connections.
7
CATH architectures
8
CATH architectures (cont.)
9
CATH hierarchical classification
- Homologous superfamily
- homologous domains identified by sequence
similarity, and structure similarity - Sequence family
- domains clustered in the same sequence
families, with sequence identitygt35
- other classification schemes SCOP, FSSP
- partial disagreement between them.
10
Growing demand for protein structures!
- PDB contains 20,868
- structures
- X-Ray and NMR have
- limitations.
WE NEED FASTER METHODS!
11
Protein Structure Prediction
- Limited to very short peptides!
12
Can known structures assist prediction?
the number of possible folds seems to be limited!
- CATH inspection more then 36,000 domains, but...
- only 800 topology groups
13
Template-based prediction (fold recognition)
II) Comparative modeling (homology modeling) -
alignment with homologous sequence of known
structure. - high sequence identity areas
similar structure - variable areas must be built
- cant be used if no sequence similarity found!
- III) Threading
- - alignment with structure sequences in fold
library - - sophisticated scoring function finds most
similar fold - - Threading aligns target sequence onto
template structure
14
What are the baselines for protein fold
recognition? McGuffin, Bryson and Jones (2001)
- Goals
- what constitutes a baseline level of success for
protein - fold recognition methods, above random guesswork?
2. can simple methods that make use of 2nd
structure information assign folds more
reliably?
- how valuable might these methods be in the rapid
- construction of a useful hierarchical
classification?
15
The methods evaluated (ordered by complexity and
runtime)
- shorten 2nd structure stringsCCCHHHHCCCEEECCHHHC
CC ? HCECH. - pairwise alignment
- scoring function also considers length of elements
16
A representative set of protein domains
- a set of 1087 domains representing different
- Sequence Families was selected from CATH.
- generate an informative file for each domain
1. gt1atx00 2. GAAaLbKSDGPNTRGNSMSGTIWVFGcPSGWNNbE
GRAIIGYacKQ 3. EEE TTS S TTSSEEEEEESS TT
EEE SSSSSEEEE 4. CEEEEEHHECEEEECCCECEEEECCCEECCE
ECEEECCEECEEEEC
17
First evaluation true positive percentage
compare true positive percentage, at a fixed 3
false positive.
run each method on all possible pairs from the
1087 set (a,b) (a,c) (a,d) ... (g,d) (g,e)
... (k,f) ... (r,s) .... 590,000 pairs
STOP! 3 false positives reached. true positive
for this method 2
1
2
2
1
3
18
We need lower,upper controls to compare with
lower control intelligent guesswork 1. randomly
assign CATH topology codes according to
frequency 2. calculate true positive, false
positive percentage
19
Optimisation of similarity scoring methods
Class pre-filter
20
- partial agreement between classification schemes
- FSSP compared with SCOP 61.1, FSSP compared
with CATH 46.7
- most accurate is method number 5 Alignment of
secondary structure - elements without additional scoring, with
27.18 true positive.
- accuracy ordering of methods doesnt correspond
to their relative complexity
21
Second evaluation CASP-like sensitivity
similarly to CASP we measure the sensitivity of
each method what is the probability of a method
correctly assigning a fold?
lower control a random proportional fold
assignment
upper control FSSP was used as a scoring method
22
Sensitivity results
- method 5 wins again 31.8 sensitivity.
- other 2nd structure based methods with small gap.
- sensitivity order of the methods true positive
percentage order.
23
Similarity trees - can we construct
classification?
Best methods similarity scores for all pairs
were clustered into a tree.
- globin-like ltgt
- casein kinase
b. immunoglobulin-like ltgt thrombin
subunit H
whole tree generally disordered
24
Conclusions
- Baseline level to be exceeded by fold
recognition methods - 27 true positive assignments allowing 3
false positive - sensitivity level of 32.
- methods which make use of 2nd structure
information - seem more accurate and sensitive than those who
dont.
- simple 2nd structure alignments alone can not
construct - reliable classification hierarchy.
- the agreement between FSSP, SCOP and CATH
- classification schemes is surprisingly low.
Recommended
CrystalGraphics Presentations





























