
Bonding in complexes of d-block metal ions - PowerPoint PPT Presentation
1 / 22
Title:
Bonding in complexes of d-block metal ions
Description:
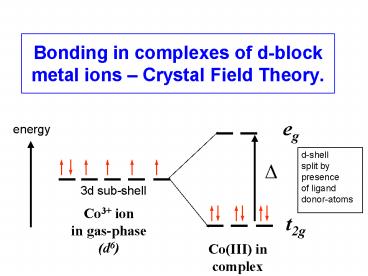
Bonding in complexes of d-block metal ions Crystal Field Theory. eg energy d-shell split by presence of ligand donor-atoms 3d sub-shell Co3+ ion – PowerPoint PPT presentation
Number of Views:95
Avg rating:3.0/5.0
Title: Bonding in complexes of d-block metal ions
1
Bonding in complexes of d-block metal ions
Crystal Field Theory.
eg
energy
d-shell split by presence of ligand donor-atoms
?
3d sub-shell
Co3 ion in gas-phase (d6)
t2g
Co(III) in complex
2
The d-orbitals
the t2g set
z
z
z
y
y
y
x
x
x
dyz
dxy
dxz
z
z
y
y
the eg set
x
x
dz2
dx2-y2
3
Splitting of the d sub-shell in octahedral
coordination
blue ligand donor atom orbitals
the egset
the t2g set
z
z
z
y
y
y
x
x
x
dyz
dz2
dx2-y2
the two orbitals of the eg set lie along
the Cartesian coordinates, and so are adjacent to
the donor atoms of the ligands, which raises the
eg set in energy
the three orbitals of the t2g set lie between the
ligand donor-atoms (only dyz shown)
4
Splitting of the d sub-shell in an octahedral
complex
energy
eg
d-shell split by presence of ligand donor-atoms
?
3d sub-shell
Co3 ion in gas-phase (d6)
t2g
Co(III) in octahedral complex
5
The crystal field splitting parameter (?)
- Different ligands produce different extents of
splitting between the eg and the t2g levels. This
energy difference is the crystal field splitting
parameter ?, also known as 10Dq, and has units of
cm-1. Typically, CN- produces very large values
of ?, while F- produces very small values.
energy
eg
eg
? 26,600 cm-1
? 15,000 cm-1
t2g
t2g
Cr(CN)63-
CrF63-
6
High and low-spin complexes
The d-electrons in d4 to d8 configurations can be
high-spin, where they spread out and occupy the
whole d sub-shell, or low-spin, where the
t2g level is filled first. This is controlled by
whether ? is larger than the spin- pairing
energy, P, which is the energy required to take
pairs of electrons with the same spin
orientation, and pair them up with the opposite
spin.
eg
Paramagnetic 4 unpaired es
eg
energy
diamagnetic no unpaired es
? gt P
? lt P
t2g
t2g
low-spin d6 electrons fill the t2g level first.
In this case the complex is diamagnetic
high-spin d6 electrons fill the whole d sub-shell
according to Hunds rule
7
High and low-spin complexes of d5 ions
For d5 ions P is usually very large, so these are
mostly high-spin. Thus, Fe(III) complexes are
usually high-spin, although with CN- ? is large
enough that Fe(CN)63- is low spin (CN- always
produces the largest ? values)
Fe(CN)63- ? 35,000 cm-1 P 19,000
cm-1
Fe(H2O)63 ? 13,700 cm-1 P 22,000
cm-1
eg
Paramagnetic 5 unpaired es
eg
energy
? gt P
? lt P
paramagnetic one unpaired e
t2g
t2g
low-spin d5 (Fe(CN)63-) electrons fill the t2g
level first. In this case the complex is
paramagnetic
high-spin d5 (Fe(H2O)63) electrons fill the
whole d sub-shell according to Hunds rule
8
High and low-spin complexes of d7 ions
The d7 metal ion that one commonly encounters is
the Co(II) ion. For metal ions of the same
electronic configuration, ? tends to increase
M(II) lt M(III) lt M(IV), so that Co(II) complexes
have a small ? and are usually high spin. The
(III) ion Ni(III) has higher values of ?, and is
usually low-spin.
Ni(bipy)33
Co(H2O)62 ? 9,300 cm-1
eg
Paramagnetic 3 unpaired es
eg
energy
? gt P
? lt P
paramagnetic one unpaired e
t2g
t2g
low-spin d7 (Ni(bipy)33) The d-electrons fill
the t2g level first, and only then does an
electron occupy the eg level.
high-spin d7 (Co(H2O)63) electrons fill the
whole d sub-shell according to Hunds rule
9
High and low-spin complexes of some d6 ions
For d6 ions ? is very large for an M(III) ion
such as Co(III), so all Co(III) complexes are
low-spin except for CoF63-.high-spin. Thus,
Fe(III) complexes are usually high-spin,
although with CN- ? is large enough that
Fe(CN)63- is low spin (CN- always produces the
largest ? values)
CoF63- ? 13,100 cm-1 P 22,000 cm-1
Co(CN)63- ? 34,800 cm-1 P 19,000
cm-1
eg
eg
Paramagnetic 4 unpaired es
energy
? gtgt P
? lt P
diamagnetic no unpaired es
t2g
t2g
low-spin d6 (Co(CN)64-) electrons fill the t2g
level first. In this case the complex is
diamagnetic
high-spin d5 (CoF63-) electrons fill the whole
d sub-shell according to Hunds rule
10
The spectrochemical series
- One notices that with different metal ions the
order of increasing ? with different ligands is
always the same. Thus, all metal ions produce the
highest value of ? in their hexacyano complex,
while the hexafluoro complex always produces a
low value of ?. One has seen how in this course
the theme is always a search for patterns. Thus,
the increase in ? with changing ligand can be
placed in an order known as the spectrochemical
series, which in abbreviated form is - I- lt Br- lt Cl- lt F- lt OH- H2O lt NH3 lt CN-
11
The spectrochemical series
- The place of a ligand in the spectrochemical
series is determined largely by its donor atoms.
Thus, all N-donor ligands are close to ammonia in
the spectrochemical series, while all O-donor
ligands are close to water. The spectrochemical
series follows the positions of the donor atoms
in the periodic table as - C N O F
- P S Cl
- Br
- I
?
very little data on P-donors may be higher than
N-donors
spectrochemical series follows arrows
around starting at I and ending at C
S-donors between Br and Cl
12
The spectrochemical series
- Thus, we can predict that O-donor ligands such
as oxalate or acetylacetonate will be close to
water in the spectrochemical series. It should be
noted that while en and dien are close to ammonia
in the spectrochemical series, 2,2bipyridyl and
1,10-phenanthroline are considerably higher than
ammonia because their sp2 hybridized N-donors are
more covalent in their bonding than the sp3
hybridized donors of ammonia.
13
The bonding interpretation of the spectrochemical
series
- For the first row of donor atoms in the periodic
table, namely C, N, O, and F, it is clear that
what we are seeing in the variation of ? is
covalence. Thus, C-donor ligands such as CN- and
CO produce the highest values of ? because the
overlap between the orbitals of the C-atom and
those of the metal are largest. For the highly
electronegative F- ion the bonding is very ionic,
and overlap is much smaller. For the heavier
donor atoms, one might expect from their low
electronegativity, more covalent bonding, and
hence larger values of ?. It appears that ? is
reduced in size because of poverlap from the
lone pairs on the donor atom, and the t2g set
orbitals, which raises the energy of the t2g set,
and so lowers ?.
14
Crystal Field Stabilization Energy (CFSE)
- When splitting of the d sub-shell occurs, the
occupation of the lower energy t2g level by
electrons causes a stabilization of the complex,
whereas occupation of the eg level causes a rise
in energy. Calculations show that the t2g level
drops by 0.4?, whereas the eg level is raised by
0.6?. This means that the overall change in
energy, the CFSE, will be given by - CFSE ?(0.4n(t2g) - 0.6n(eg))
- where n(t2g) and n(eg) are the numbers of
electrons in - the t2g and eg levels respectively.
15
Calculation of Crystal Field Stabilization Energy
(CFSE)
- The CFSE for some complexes is calculated to be
- Co(NH3)63 Cr(en)33
energy
eg
eg
t2g
t2g
? 22,900 cm-1 ? 21,900 cm-1 CFSE
22,900(0.4 x 6 0.6 x 0) CFSE 21,900(0.4 x
3 0.6 x 0) 54,960 cm-1 26,280 cm-1
16
Crystal Field Stabilization Energy (CFSE) of d5
and d10 ions
- The CFSE for high-spin d5 and for d10 complexes
is calculated to be zero - Mn(NH3)62 Zn(en)33
energy
eg
eg
t2g
t2g
? 22,900 cm-1 ? not known CFSE
10,000(0.4 x 3 0.6 x 2) CFSE ?(0.4 x 6
0.6 x 4) 0 cm-1 0 cm-1
17
Crystal Field Stabilization Energy (CFSE) of d0
to d10 M(II) ions
- For M(II) ions with the same set of ligands, the
variation of ? is not large. One can therefore
use the equation for CFSE to calculate CFSE in
terms of ? for d0 through d10 M(II) ions (all
metal ions high-spin) - Ca(II) Sc(II) Ti(II) V(II) Cr(II)
Mn(II) Fe(II) Co(II) Ni(II) Cu(II)
Zn(II) - d0 d1 d2 d3
d4 d5 d6 d7 d8
d9 d10 - CFSE 0 0.4? 0.8? 1.2? 0.6? 0
0.4? 0.8? 1.2? 0.6? 0 - This pattern of variation CFSE leads to greater
stabilization in the complexes of metal ions with
high CFSE, such as Ni(II), and lower
stabilization for the complexes of M(II) ions
with no CFSE, e.g. Ca(II), Mn(II), and Zn(II).
The - variation in CFSE can be compared with the log
K1 values for EDTA - complexes on the next slide
18
Crystal Field Stabilization Energy (CFSE) of d0
to d10 M(II) ions
Ni2
double- humped curve
Ca2
Mn2
Zn2
19
Log K1(EDTA) of d0 to d10 M(II) ions
CFSE
double- humped curve
Zn2
Mn2
rising baseline due to ionic contraction
Ca2
20
Log K1(en) of d0 to d10 M(II) ions
CFSE
double- humped curve
Zn2
rising baseline due to ionic contraction
Ca2
Mn2
21
Log K1(tpen) of d0 to d10 M(II) ions
double- humped curve
Zn2
Mn2
tpen
Ca2
22
The Irving-Williams Stability Order
- Irving and Williams noted that because of CFSE,
the log K1 values for virtually all complexes of
first row d-block metal ions followed the order - Mn(II) lt Fe(II) lt Co(II) lt Ni(II) lt Cu(II) gt
Zn(II) - We see that this order holds for the ligand
EDTA, en, and TPEN on the previous slides. One
notes that Cu(II) does not follow the order
predicted by CFSE, which would have Ni(II) gt
Cu(II). This will be discussed under Jahn-Teller
distortion of Cu(II) complexes, which leads to
additional stabilization for Cu(II) complexes
over what would be expected from the variation in
CFSE.
Recommended
CrystalGraphics Presentations





























