
Defining what a tree means - PowerPoint PPT Presentation
1 / 37
Title:
Defining what a tree means
Description:
methods for going from the data to a tree can be put into two general categories: ... ( also inferior to ML methods, which are not much more computationally ... – PowerPoint PPT presentation
Number of Views:55
Avg rating:3.0/5.0
Title: Defining what a tree means
1
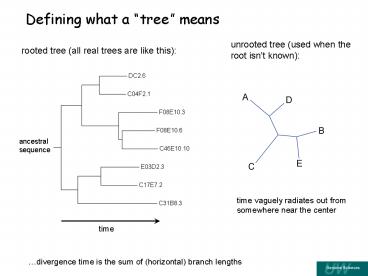
Defining what a tree means
unrooted tree (used when the root isnt known)
rooted tree (all real trees are like this)
ancestral sequence
time vaguely radiates out from somewhere near the
center
divergence time is the sum of (horizontal)
branch lengths
2
(No Transcript)
3
(No Transcript)
4
(No Transcript)
5
(No Transcript)
6
(No Transcript)
7
(No Transcript)
8
(No Transcript)
9
(No Transcript)
10
Toms favorite tree (vertebrate Cyp450)
11
A tree has topology and distances
Note - topologically, these are the SAME tree. In
general, two trees are the same if they can be
inter-converted by branch rotations.
12
The number of tree topologies grows extremely fast
3 leaves 3 branches 1 internal node 1 topology (3
insertions)
4 leaves 5 branches 2 internal nodes 3 topologies
(x3) (5 insertions)
In general, an unrooted tree with N leaves
has 2N 3 branches N 2 internal nodes O(N!)
topologies
5 leaves 7 branches 3 internal nodes 15
topologies (x5) (7 insertions)
13
There are many rooted trees for each unrooted tree
For each unrooted tree, there are 2N - 3 times as
many rooted trees, where N is the number of
leaves ( internal branches 2N 3).
14
Trees can be enormously complex
This tree has 203 7-pass chemosensory protein
sequences and could adopt gtgtgt10100 topologies.
Even a tree of 20 sequences could adopt 2 x 1020
topologies.
Note that this does NOT include branch length
differences!
15
Duplication and Divergence
- Full gene duplication can occur in two patterns
- 1) speciation (orthology)
- 2) duplication within species (paralogy)
- Gene duplication within a species probably
occurs only during aberrant DNA rearrangements. - Immediately after duplication, the two copies of
a gene are fully redundant and one often mutates
to non-functionality and fixes in the population
(pseudogene). - Some of the time (unclear how often) the two
copies diverge to become functionally distinct
enough that each function is under negative
selection and both are retained.
16
Orthology and paralogy trees
17
How duplications can arise
2N ? 4N (e.g. failure in mitosis)
1) genome duplication
2) unequal crossing-over
3) replication slippage
4) transposon hops run amock
5) other events involving repair of double strand
breaks
18
To reconstruct species phylogeny
- sequence orthologs from each species
- construct gene tree it probably corresponds to
the species tree (question assuming you
construct the gene tree correctly what could
violate this? TWO answers) - do this for more than one gene to be sure you
should get nearly the same tree every time.
19
Species phylogeny (and a footnote on
humanocentrism)
This tree is amazingly misleading
correct tree
Not to mention 1) the straight line to humans,
2) leaving out gt90 of all species, including
three hominoids bonobo, siamang, gibbon, and 3)
the fact that WE are an African ape.
20
(No Transcript)
21
How do you make a tree from data?
- data typically takes the form of either a
multiple sequence alignment or a set of all
pairwise sequence alignments. - methods for going from the data to a tree can be
put into two general categories - - Distance Matrix methods (e.g. UPGMA,
Neighbor-Joining) - - Tree-enumerating methods (e.g. parsimony,
maximum- likelihood) - in theory, the tree-enumerating methods are
superior, but they are not always practical.
22
Distance matrix methods
- methods based on a set of pairwise distances (no
multiple alignment needed, though pairwise
distances often come from one). - all methods (in essence) build a tree that tries
to best match the distances. - usual standard for best match is the least
squares of the tree distances compared to the
real pairwise distances
Let Dm be the matrix distances and Dt be the
tree distances. Find the tree (an internally
consistent set of Dt values) that minimizes
23
Example distance matrix (ID) and tree
EGL-2 dEag rEag UNC-103 dErg HERG EGL-2 0.000 0.2
76 0.353 0.542 0.547 0.525 dEag 0.276 0.000 0.305
0.512 0.501 0.508 rEag 0.353 0.305 0.000 0.533 0.5
15 0.510 UNC-103 0.542 0.512 0.533 0.000 0.274 0.2
55 dErg 0.547 0.501 0.515 0.274 0.000 0.263 HERG 0
.525 0.508 0.510 0.255 0.263 0.000
24
Least-squares solution
- There are methods that directly solve the
least-squares problem - However, they are computationally slow and
rarely used. - Fortunately, there are more direct
approximations that work remarkably well, most
notably Neighbor-Joining. - The direct methods use various types of
sequential clustering.
25
Sequential clustering approach
26
Neighbor-Joining Algorithm (NJ)
Essentially as on previous slide, but correction
for distance to other leaves is made.
Specifically, for two leaves i and j, we denote
the set of all other leaves as L, and the size of
that set as , and we compute the corrected
distance Dij as
heres an intuitive rationale (consider
clustering the first two leaves)
27
Neighbor-Joining corrects for different rates of
evolution on branches
these two branches have changed faster
The corrective terms change the distance from
leaf 1 and 3 so that they are clustered before
leaf 1 and 2 (and similarly for leaves 2 and 4).
28
Parsimony method
- intuitively appealing find the tree that can
explain the observed sequences with the smallest
number of changes. - BUT requires enumeration of tree topologies, so
not feasible except for large data sets. (also
inferior to ML methods, which are not much more
computationally intensive).
1 AAG 2 AAA 3 GGA 4 AGA
29
Maximum Likelihood Method
- First developed by Joe Felsenstein here at UW.
- Formulate a specific model of evolution, with
rates and probabilities of various changes. - Assess the likelihood of the data set given a
particular tree topology and branch length set
(and your model). - Use a tree-rearrangement/hill-climbing method to
move from reasonably good trees toward the best
tree. - Even more computationally demanding than
parsimony, but probably the best method
especially when statistical analysis is critical.
30
Tree topology enumeration is very problematic
3 leaves 3 branches 1 internal node 1 topology (3
insertions)
4 leaves 5 branches 2 internal nodes 3 topologies
(x3) (5 insertions)
In general, a tree with N leaves has 2N 3
branches N 2 internal nodes O(N!) topologies
5 leaves 7 branches 3 internal nodes 15
topologies (x5) (7 insertions)
31
Speed up in topology finding
- Used in both parsimony and maximum-likelihood
methods. - Start with an NJ distance tree, assuming that it
approximates the correct tree. - Assess the likelihood of the tree, then test
multiple simple rearrangements of the tree
looking for better likelihoods. - Repeat until nothing gets better.
32
Topology statistics the bootstrap
- we can say that the data are most consistent
with a particular tree (with one or another
method) but how confident are branch points? - for example
33
Bootstrap and jack-knife calculation (resampling)
- General principal is to stress the data
repeatedly and recompute the tree each time,
looking for robust features. - Jack-knife drop each of N sequences from the
alignment and recompute the resulting N trees,
testing whether they are compatible with
original. - Bootstrap recompute pairwise distances from a
random sample of alignment columns (with
replacement). Recompute the tree for each new
distance set and see how often a particular tree
branch is positioned the same.
34
Bootstrap (cont.)
real alignment
Compute relative distances among pairs by
comparing scores (not discussed in class).
Similar resampling and distance derivation done
for each sequence pair, then a new tree
calculated. Repeat ad nauseum.
EGL-2 dEag rEag UNC-103 dErg HERG EGL-2 0.000 0.2
76 0.353 0.542 0.547 0.525 dEag 0.276 0.000 0.305
0.512 0.501 0.508 rEag 0.353 0.305 0.000 0.533 0.5
15 0.510 UNC-103 0.542 0.512 0.533 0.000 0.274 0.2
55 dErg 0.547 0.501 0.515 0.274 0.000 0.263 HERG 0
.525 0.508 0.510 0.255 0.263 0.000
35
The molecular clock concept
- divergence distance vs. time NOT the same!
- molecular clock is common default hypothesis
for translating distance into time (assume that
divergence time). - known to be violated whenever sufficiently broad
groups are considered. - however, frequently approximately valid for a
particular sequence family over relatively short
times (e.g. most proteins in primates).
36
Things that tend to invalidate the molecular clock
- differential changes in generation time on some
branches. - differential changes in selective constraints on
some branches (extreme example would be positive
selection). - depth of divergence - though correctable unless
distances are too long.
37
CAUTIONARY NOTES ON VARIOUS METHODS
- All tree building methods depend strongly on
high quality alignments. To the degree that
alignments are problematic (deep divergence) the
trees are problematic. Bootstrap and other
methods do NOT fully account for this. - For good alignments of sequences that are not
excessively divergent, all tree-building methods
should give fairly similar trees. (If they dont,
be skeptical) - Trees that LOOK very different often are not
you may have to reroot the tree and rotate tree
branches to get two trees to look similar to the
human eye.
Recommended
CrystalGraphics Presentations





























