
V11 Modelling genetic networks by boolean networks - PowerPoint PPT Presentation
1 / 43
Title:
V11 Modelling genetic networks by boolean networks
Description:
... xij X, where the arguments xj are limited to the parent vertices ... degree of conservation. by a four-way species comparison. Assemble with Arachne program ... – PowerPoint PPT presentation
Number of Views:141
Avg rating:3.0/5.0
Title: V11 Modelling genetic networks by boolean networks
1
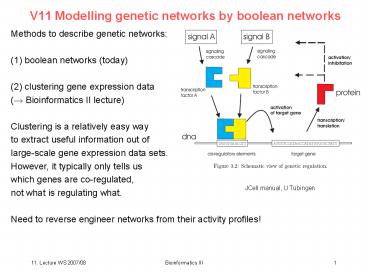
V11 Modelling genetic networks by boolean networks
Methods to describe genetic networks (1)
boolean networks (today) (2) clustering gene
expression data (? Bioinformatics II
lecture) Clustering is a relatively easy way to
extract useful information out of large-scale
gene expression data sets. However, it typically
only tells us which genes are co-regulated, not
what is regulating what. Need to reverse
engineer networks from their activity profiles!
JCell manual, U Tübingen
2
Boolean networks
Boolean networks allow dynamic modelling of
synchronous interactions between vertices in a
network. They belong to the simplest models that
possess some of the biological and systemic
properties of real gene networks. In Boolean
logic, a Boolean variable x is a variable that
can assume only two values. The values are
denoted usually as 0 and 1, and correspond to the
logical values true and false. The logic
operators and, or, and not are defined to
correspond to the intuitive notion of
truthfulness and composition of those operators.
A Boolean function is a function of Boolean
variables connected by logic operators.
3
Boolean networks
A Boolean network is a directed graph G(X,E),
where the vertices, xi ? X, are Boolean
variables. To each vertex, xi, is associated a
Boolean function, b(xi1, xi2, , xil) , l ? n,
xij ? X, where the arguments xj are limited to
the parent vertices of xi in G. Together, at
any given time, the values of all vertices
represent the state of the network, given by the
vector S(t) (xi1(t), xi2(t), ,
xilt)). For gene networks, the vertex variables
correspond to levels of gene expression,
discretized to either 0 or 1. The Boolean
functions at the vertices model the aggregated
regulation effect of all their parent vertices.
The states of all nodes are updated at the same
time according to their respective Boolean
functions
4
Boolean networks
The transitions of all states together correspond
to a state transition of the network from S(t) to
the new network state, S(t1). A series of
state transitions is called a trajectory. Since
there is a finite number of network states, all
trajectories are periodic. This simply follows
from the fact that as soon as one state is
visited a second time, the trajectory will take
exactly the same path as for the first time.
The repeating parts of the trajectories are
called attractors, and can be one or more states
long. All the states leading to the same
attractor are the basin of attraction for this
attractor.
5
Reverse engineering Boolean Networks
Clustering is a relatively easy way to extract
useful information out of large-scale gene
expression data sets. However, it typically only
tells us which genes are co-regulated, not which
gene is regulating what other gene(s). The goal
in reverse engineering Boolean networks is to
infer both the underlying topology (i.e. the
edges in the graph) and the Boolean functions at
the vertices from observed gene expression data.
The actual observed data can come either from
gene expression experiments conducted at
different time intervals or when the expression
of various genes is perturbed. For time-course
data, measurements of the gene expressions at two
consecutive time points simply correspond to two
consecutive states of the network, S(i) and
S(i1).
6
Intergenic interaction matrix M
Perturbation data come in pairs, which can be
thought of as the input/output states of the
network, Ii/Oi, where the input state is the one
before the perturbation and the output the one
after it. Given the observations of the states
of a Boolean network, in general many networks
may be constructed that are consistent with that
data. Hence the solution network is ambiguous.
There are several variants of the reverse
engineering problem (a) finding one network
consistent with the data, (b) finding all
networks consistent with the data, and (c)
finding the 'best' network consistent with the
data (according to some pre-specified criteria).
The first task is the simplest one and efficient
algorithms exist.
7
Reverse engineering of boolean networks
The reverse engineering problems are intimately
connected to the amount of empirical data
available. Obviously, the inferred network will
be less ambiguous the more data points are
available. The amount of data needed to
completely determine a unique network is known as
the data requirement problem in network
inference. The amount of data required depends
on the sparseness of the underlying topology and
the type of Boolean functions allowed. This can
be understood intuitively. A network with few
connections may be defined with few data points.
In the worst case, the deterministic inference
algorithms need on the order of m 2n
transition pairs of data (experimental data
points) to infer a densely connected Boolean
Network with general Boolean functions at the n
vertices.
Aracena Demongeot, Acta Biotheoretica 52, 391
(2004)
8
Intergenic interaction matrix M
Since introducing the detection of gene
expression by microarrays, a major problem has
been the estimation of the intergenic interaction
matrix M. In V10, the entries of M were either 0
or 1. Today, we allow the matrix element mij of
the interaction matrix M to be - positive if gene
Gj activates gene Gi - negative if gene Gj
inhibits gene Gi - equal to 0 if gene Gj and
gene Gi have no interaction.
9
simulating the dynamics of regulatory networks
Given the interaction matrix M, the change of
state xi of gene Gi between t and t 1 obeys a
threshold rule
where H is the Heavyside function H(y) 1 if
y ? 0 and H(y) 0 if y lt 0, and the bis are
threshold values. In the case of small
regulatory genetic systems, the knowledge of such
a matrix M makes it possible to know all possible
stationary behaviors of the organisms having the
corresponding genome.
Aracena Demongeot, Acta Biotheoretica 52, 391
(2004)
10
Example
In the genetic regulatory network which rules
Arabidopsis thaliana flower morphogenesis
(right), the interaction matrix is a (11,11)
matrix with only 22 non zero coefficient.
Below A fixed configuration (attractor) of its
Boolean dynamics that is obtained from
propagating xi(t).
Mendoza, Alvarez-Buylla, JCB, 1998
11
Reverse engineering of the interaction matrix
For each genetic regulatory network, we can
define an interaction graph built from the
interaction matrix M by drawing an edge (resp.
-) between the vertices representing the genes j
and i, iff mij gt 0 (resp. lt 0). To calculate the
mijs, we can either determine the s-directional
correlation ?ij(s) between the state vector xj(t
s)t ? C of gene j at time t s and the state
vector xi(t)t ? C of gene i at time t , t
varying during the cell cycle C of length K C
and corresponding to the observation time of
the bio-array images
Aracena Demongeot, Acta Biotheoretica 52, 391
(2004)
12
interaction matrix
and then take
where ? is a de-correlation threshold. Alternativ
ely, one may identify the system with a Boolean
neural network. When it is impossible to obtain
all the coefficients of M in this manner (either
from the literature or from such calculations),
it may be possible to complete M by appyling an
heuristic approach.
Aracena Demongeot, Acta Biotheoretica 52, 391
(2004)
13
estimation of interaction values
We may randomly choose the missing coefficients
by considering - the connectivity coefficient
K(M) I / N, the ratio between the number I of
interactions and the number N of genes, and - the
mean inhibition weight I(M) R / I , the ratio
between the number of inhibitions R and I. For
many known operons and regulation networks, K(M)
is between 1.5 and 3, and I(M) between 1/3 and
2/3. If M is structurally stable, then the
random estimation of M can be used to obtain an
approximate estimation on the control mechanisms
of the regulatory network.
Aracena Demongeot, Acta Biotheoretica 52, 391
(2004)
14
Monod and Jacob (1961) first proposed that
complex networks of gene interactions regulate
cell differentiation. The first formal models of
genetic regulation of cell differentiation
anticipated that real biological genetic networks
would be too complex to be analyzed without the
use of formal mathematical and/or computational
tools. However, because the early models made
some assumptions that were biologically
unrealistic, experimentalists largely ignored
them. Relatively complete genetic descriptions of
developmental programs are now available in
several model organisms, providing the necessary
inputs for developing biologically realistic
dynamic models of gene regulatory networks in
cell differentiation. Such models should aid at
building a formal framework for studies of
developmental mechanisms and their evolution.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
15
methods
The model is discrete. N is the number of genes
involved in the network, Xn is a vector with
expression state for each gene in a space of N
dimensions, representing the network state after
n iterations
where xi(n) represents the state of expression of
the gene i at the iteration n. We then write
indicating that the state at the iteration (n
1) is determined by the state at the previous
iteration. Each node, except SEP (redundant
SEP1, SEP2, and SEP3 genes), stands for the
activity of a single gene involved in floral
organ fate determination. Most nodes could
assume three levels of expression (on, 1 or 2,
and off, 0) to enable different activation
thresholds when experimental data was available.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
16
functions of genes inferred from experiments
FLOWERING LOCUS T Double embryonic flower1 (emf1)
flowering locus t (ft) mutants do not develop
embryonic flowers typical of emf1 single mutants,
suggesting that the lack of FT activity
suppresses the emf1 phenotype because EMF1
represses FT. LFY Double mutants of the weak
emf1-1 allele and lfy-1 bear lfy-like flowers,
suggesting that, for this trait, lfy is
epistatic. These genes have antagonistic
activities hence, we infer that LFY is repressed
by EMF1. TFL1 In emf1-2 tfl1 double mutants, the
emf1-2 mutation is epistatic. As these two genes
do not have opposite functions, this result
suggests that EMF1 protein is needed for TFL1
activity in wild-type Arabidopsis. ....
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
17
logical rules
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
18
logical rules
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
19
logical rules
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
20
network architecture
Figure 4. Gene Network Architecture for the
Arabidopsis Floral Organ Fate Determination.
Network nodes represent active proteins of
corresponding genes, and the edges represent the
regulatory interactions between node pairs
(arrows are positive, and blunt-end lines are
negative). Dashed lines are hypothetical
interactions for which there is no experimental
support (see logical rules). The network includes
F-box proteins (UFO), membrane bound signaling
molecules (TFL1 and FT), cofactors involved in
transcriptional regulation (EMF1 and LUG),
chromatin remodeling proteins (CLF), and
transcription factors (all others). Interactions
have been confirmed to be direct transcriptional
regulations in a few cases (LFY on AG, LFY on
AP1), and the rest can either be direct or
indirect and can be transcriptional or other.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
21
states of the system
The system has a finite number of possible
initial conditions equal to 139,968, and each
one is represented by a vector of dimension 15 in
which each column corresponds to the expression
state of each network node at initial conditions
in the following order FT EMF1 TFL1 LFY FUL AP1
AP3 PI AG UFO WUS AP2 SEP LUG CLF. The vector
of 15 entries that keeps track of the activity
level of each node describes the system at each
time point. We updated the state of each node
synchronously. Starting on each initial
condition the network is iterated until it
reached an attractor. The 139,968 initial states
converge to only 10 stable attractors. All of
them are fixed point attractors in which the
activity level of all genes remains the same as
in the previous iteration, see table 1.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
22
simulated gene expression levels
These steady gene states (Table 1) predicted by
the model coincide with the gene expression
profiles that have been documented experimentally
in cells of wild-type Arabidopsis inflorescence
meristems and floral organ primordia. For
example, in the Infl steady states, floral
meristem identity genes (LFY, AP1, and AP2) and
floral organ identity genes (AP1, AP2, AP3, PI,
SEP, and AG) are off, whereas the inflorescence
identity genes (EMF1 and TFL1) are on.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
23
attractors
The size of the basins of attraction may indicate
how stable each morphogenetic response is and
which genes are critical to attain each cell fate
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
24
petunia gene network
Deciphering network architectures underlying cell
differentiation is a first step toward
understanding the mechanisms that rule
conservation and variation in morphological
traits. Although most flowering species have an
overall conserved plan of floral organ
determination, mutations have revealed some
important variations. In the case of petunia, the
overall network of cell fate determination during
flower organ development seems to be conserved
with respect to Arabidopsis. However, mutant
analyses have suggested some differences. Our
simulation results suggest that an architecture
like the one proposed here for the Arabidopsis
network that includes a duplicated AP3 would
yield the gene expression patterns observed for
the wild type and a single AP3-like gene mutant
and provides a prediction for the double
loss-of-function mutations of AP3-like genes in
petunia.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
25
comparison arabidopsis - petunia
Figure 5. Arabidopsis and Petunia Wild Type
(left), ap3 Mutant (right) Flowers, and
Corresponding Network Models. Single petunia
mutant for PhDEF is shown in the top part of (B)
and a scheme of the predicted double mutant for
PhDEF and PhTM6 is shown below. Arabidopsis is
shown in (A). The networks indicate which nodes
were turned off (yellow) to simulate mutants. The
Arabidopsis orthologs of the cloned petunia genes
are as follows FLORAL BINDING PROTEIN26 (FBP26)
is an AP1 ortholog, PhDEF (formerly known as
GREEN PETALS) is an AP3 and DEFICIENS (DEF from
A. majus) ortholog, PhGLO1 (FBP1) and PhGLO2
(PETUNIA MADS BOX GENE2 pMADS2) are PI and
GLOBOSA (GLO from A. majus) orthologs, pMADS3 is
an AG ortholog, PhAP2A is an AP2 ortholog, FBP2
and FBP5 are SEP orthologs, PhCLF1 and PhCLF2 are
CLF orthologs, and PETUNIA HYBRIDA TM6 (PhTM6) is
a paleoAP3 gene.
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
26
comparison arabidopsis - petunia
Espinosa-Soto et al. The Plant Cell 16, 2923
(2004)
27
Control of Gene Expression
A bacterial cell lives in direct contact with its
environment. Its chemical composition may
dramatically change from one moment to the
other. Consider bacteria growing either on
lactose or tryptophan. Fig. 2.16 Lactose
di-saccharide from glucose galactose oxidatio
n provides cells with metabolic
intermediates and energy. First step of
lactose degradation (catabolism) hydrolysis
of the bond joining the 2 sugars
by ?-galactosidase
Karp Cell Mol. Biol.
28
Transfer from minimal medium to lactose medium
When bacterial cells are grown in a minimal
medium, they dont need ?-galactosidase and
contain lt 5 copies and only 1 copy of its
mRNA. What happens when the cells are
transferred to a lactose medium?
Karp Cell Mol. Biol.
29
lac Operon an inducible operon
Inducible operon presence of substance
(lactose) induces transcription of the
structural genes. lac operon contains 3 tandem
structural genes z gene encodes
?-galactosidase y gene encodes galactoside
permease, a protein that promotes entry of
lactose into the cell a gene encodes
thiogalactoside acetyltransferase
Karp Cell Mol. Biol.
30
positive control by cyclic AMP
Repressors, such as those of the lac and trp
operons, exert their influence by negative
control. lac operon is also under positive
control, the glucose effect. If bacterial
cells are supplied with glucose (as well as with
other substances such as lactose or galactose),
the cells catabolize the glucose and ignore the
other compounds. ? glucose in the medium
suppresses the production of various catabolic
enzymes, such as ?-galactosidase, needed to
degrade the other substrates. In 1965, cAMP was
deteced in E.coli. The higher the glucose
concentration, the lower the cAMP concentration.
When adding cAMP to the medium in the presence of
glucose, the catabolic enzymes that were normally
absent were suddenly synthesized by the
cell. cAMP binds to CRP. The cAMP-CRP complex
recognizes and binds to a specific site in the
lac control region. The presence of bound CRP
changes the DNA conformation and allows RNA
polymerase to transcribe the lac operon.
Karp Cell Mol. Biol.
31
positive control by cyclic AMP
Fig. 12.27
Karp Cell Mol. Biol.
32
Growth on Trp medium
Trp is required for protein synthesis. If no Trp
is available in the medium, the bacterium must
expend energy synthesizing this amino acid ?
cells contain enzymes and corresponding mRNA of
Trp-synthesis pathway. If Trp becomes available
in the medium, the cells no longer have to
synthesize their own Trp. Within a few minutes,
the production of the enzymes of the Trp pathway
stops. In the presence of Trp, the genes encoding
these enzymes are repressed.
Karp Cell Mol. Biol.
33
trp operon
In a repressible operon, the repressor is
unable to bind to the operator DNA itself.
Karp Cell Mol. Biol.
34
eukaryotic gene expression PEPCK
model case gene that codes for
phosphoenolpyruvate carboxykinase (PEPCK). This
enzyme is one of the key enzymes of
gluconeogenesis, the metabolic pathway that
converts pyruvate to glucose. The enzyme is
synthesized in the liver when glucose levels are
low, e.g. when considerable time has passed since
your last meal. Synthesis drops sharply after
carbohydrate-rich meal.
Level of synthesis of PEPCK mRNA is controlled by
a variety of transcription factors, including
several hormone receptors that are involved in
regulating carbohydrate metabolism. To
understand the regulation of PEPCK gene
expression, we need to (1) unravel the functions
of the numerous DNA regulatory sequences that
residue upstream from the gene itself (2)
identify the transcription factors that bind
these sequences, and (3) identify the signalling
pathways that activate the machinery responsible
for selective gene expression.
Karp Cell Mol. Biol.
35
eukaryotic gene expression PEPCK
Fig. 12.32 TATA box followed by
core promoter site of assembly of a
pre-initiation complex consisting of RNA
polymerase II and a number of general TFs CAAT
GC boxes bind global TFs such as NF1 and SP1
both are typically located 100 150 bp upstream
? proximal promoter elements
Karp Cell Mol. Biol.
36
Responsive elements from PEPCK gene
various hormones affect the expression of PEPCK
gene insulin, thyroid hormone, glucagon,
glucocorticoid. All of the act by means of
specific TFs that bind to the DNA. Fig. shows
responsive elements.
Karp Cell Mol. Biol.
37
Activation of transcription
For example, let us focus on glucocorticoids, a
group of steriod hormones (e.g. cortisol) that
are synthesized in response to stress. Fig.
12.34
Karp Cell Mol. Biol.
38
Conservation of regulatory elements?
Nature 423, 241 (2003)
39
Comparative genome analysis
Compare sequences of Saccharomyces paradoxus, S.
mikatae, S. bayanus, with S. cerevisae. The
three new yeast species have sufficient sequence
similarity to S. cerevisiae to allow orthologous
regions to be aligned reliably, but sufficient
sequence divergence to allow many functional
elements to be recognized by their greater
degree of conservation by a four-way species
comparison. Assemble with Arachne program Align
4 genomes.
Nature 423, 241 (2003)
40
Conservation of the Gal4-binding site
We first studied the binding site for one of the
best-studied transcription factors, Gal4, whose
sequence motif is CGGn(11)CCG (which contains 11
unspecified bases). Gal4 regulates genes
involved in galactose metabolism, including the
GAL1 and GAL10 genes, which are divergently
transcribed from a common intergenic region.
The Gal4 motif occurs three times in this
intergenic region, and all three instances show
perfect conservation across the four species.
In addition, there is a fourth experimentally
validated binding site for Gal4 that differs from
the consensus by one nucleotide in S. cerevisiae.
This variant site is also perfectly preserved
across the species.
Nature 423, 241 (2003)
41
Conservation of the Gal4-binding site
We then examined the frequency and conservation
of Gal4-binding sites across the aligned genomes.
In S. cerevisiae, the Gal4 motif occurs 96 times
in intergenic regions and 415 times in genic
(protein-coding) regions. The motif displays
certain marked conservation properties (1)
occurrences of the Gal4 motif in intergenic
regions have a conservation rate (proportion
conserved across all four species) that is about
fivefold higher than for equivalent random
motifs. (2) intergenic occurrences of the Gal4
motif are more frequently conserved than genic
occurrences. By contrast, random motifs are less
frequently conserved in intergenic regions than
in genic regions, reflecting the lower overall
level of conservation in intergenic regions.
Thus, the relative conservation rate in
intergenic compared with genic regions is about
11-fold higher for Gal4 than for random motifs.
(3) the Gal4 motif shows a higher conservation
rate in divergent compared with convergent
intergenic regions (those that lie upstream
compared with downstream of both flanking genes)
no such preferences are seen for control motifs.
These three observations suggest various ways to
discover motifs based on their conservation
properties.
Nature 423, 241 (2003)
42
43
Assign function
Assign candidate functions to these discovered
motifs by the genes adjacent to conserved
occurrences of the motif with known gene
categories. Test for Gal4 motif. Given the
biological role of Gal4, we considered the set of
genes annotated to be involved in carbohydrate
metabolism (126 genes according to the Gene
Ontology classification) with the set of genes
that have a Gal4-binding motif upstream. The
intergenic regions adjacent to carbohydrate
metabolism genes comprise only 2 of all
intergenic regions, but 7 of the occurrences of
the Gal4 motif in S. cerevisiae and 29 of the
conserved occurrences across the four species. ?
suggests that a function of the Gal4 motif could
be inferred from the function of the genes
adjacent to its conserved occurrences. Such
putative functional assignments can be useful in
directing experimentation for understanding the
precise function of a motif. Such
considerations indicate that it should be
possible to use comparative analysis, such as
explored here for yeast, to identify directly
many functional elements in the human genome that
are common to mammals. More generally,
comparative analysis offers a powerful and
precise initial tool for interpreting genomes.
Nature 423, 241 (2003)
Recommended
CrystalGraphics Presentations





























