
Simplest case: stretches of very highly conserved sequence
1 / 28
Title:
Simplest case: stretches of very highly conserved sequence
Description:
'Phylogenetic Shadowing of Primate Sequences to Find. Functional Regions of the Human Genome ... primates, including human sequence. ... –
Number of Views:47
Avg rating:3.0/5.0
Title: Simplest case: stretches of very highly conserved sequence
1
Simplest case stretches of very highly
conserved sequence
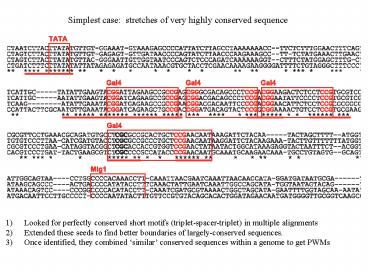
- Looked for perfectly conserved short motifs
(triplet-spacer-triplet) in multiple alignments - Extended these seeds to find better boundaries of
largely-conserved sequences. - Once identified, they combined similar
conserved sequences within a genome to get PWMs
2
If species are too closely related, many
sequences may be conserved by chance and
therefore its difficult to identify functionally
conserved sequences
Boffellii et al. 2003 Phylogenetic Shadowing of
Primate Sequences to Find Functional Regions of
the Human Genome
3
If species are too closely related, many
sequences may be conserved by chance and
therefore its difficult to identify functionally
conserved sequences
Boffellii et al. 2003 Phylogenetic Shadowing of
Primate Sequences to Find Functional Regions of
the Human Genome
- Alignments of orthologous sequence from 18 Old
World and New World - primates, including human sequence.
- They generated sequence over specific loci from
18 Old World primates, - New World primates, and hominoids.
- Multiple alignments using Clustal W
- Estimated mutation rates using HKY model of
mutation - Generated a species tree using a maximum
likelihood method - Calculated the log likelihood of fast mutation
rate vs. slow mutation rate
4
Relative likelihood of fast evolution model
vs. slow evolution model
This method is especially useful for identifying
primate-specific genes regulatory regions.
5
Transfection assays tested predicted conserved
(functional) and nonconserved (nonfunctional)
regions
Conserved regions
Unconserved regions
6
This group looked for long-range enhancers, not
based on predicted enhancers that are conserved,
but by defining syntenic blocks of CNSs (in
human-mouse-chicken human-mouse-frog). Idea is
that there will be linkage disequilibrium for
linked regulatory elements.
7
Whole-genome alignments show breaks in synteny
Breaks in synteny can be tolerated if they dont
disrupt important linkages. Here search
alignments of human-mouse-chicken/human-mouse-frog
for blocks of conservation linked beyond that
expected.
8
The above approaches have focused on using
similarity/conservation to identify important
regions of the genome A large focus in
genomics is understanding the differences in
genome sequences and what accounts for the vast
diversity in phenotypes within a population.
Analysis of single nucleotide polymorphisms (SNP)
within populations, Analysis of variations in
gene expression within and between
populations, Analysis of quantitative trait loci
(QTLs) accounting for differences in gene
expression.
9
Connecting phenotype to genotype
-- Large variations in size, shape, health, etc
in human populations -- Much of that variation
has to do with disease susceptibility -- A major
goal of genetics (and now genomics) is
understanding the consequences of genetic
variation. 1200 disease-associated genes
known, mostly by positional cloning mapping
studies. Done by linkage analysis pattern of
marker inheritance in families with heritable
diseases
A major force in genomics is to identify and
annotate SNPs in human populations. Lots of
HapMap Consortia working toward this goal. gt 5
million SNPs estimated with allele frequencies
gt5 in the human population. 4 million SNPs
estimated with allele frequencies between 1-5 in
the human population.
10
(No Transcript)
11
Each base-pair position on human chromosome 21 is
interrogated 8 times (4 in forward 4 in
reverse orientations)
GGAGATGAGTTCGATTACTCTTAGG
GGAGATGAGTTCAATTACTCTTAGG
GGAGATGAGTTCTATTACTCTTAGG
GGAGATGAGTTCCATTACTCTTAGG
1.7 x 108 oligos total on eight Affy wafers were
used to identify SNPs on human Chromosome 21 from
21 different individuals.
12
Patil et al identified 35,989 SNPs on 20
independent copies of human Ch. 21 11,000 SNPs
(32) occurred once in their population lt 10
allele frequency (neutral model predicted 42 of
genes would have single SNPs) SNPs are not
independent of one another and are often
linked. Haplotype blocks stretches of
contiguous, linked SNPs -- used 24,000 SNPs to
construct haplotype blocks
13
Each row single SNP Each column Ch 21 Blue
major allele Yellow minor allele
Much of the chromosomal variation is explained
with relatively limited haplotype
diversity. 80 of haplotype structure can be
captured with only 10 of the SNPs in that
block (need only 2SNPs to type) Haplotype
length can vary from a few kb to mega bases.
14
Assayed 1,586,383 SNPs in 71 Americans (of
European, African, Asian descent)
15
1.3 of their SNPs are in coding regions .
therefore, most of these SNPs are in noncoding
regions.
18 of SNPs represent private SNPs observed in
only one population (mostly the African-American
population). Higher frequency of rare alleles in
the African-American population However, most of
the variation (SNPs) they observed were not
population-specific.
Regions of strong linkage disequilibrium (LD)
occur in regions with low recombination rates.
16
(No Transcript)
17
Phenotypic variation (including disease
susceptibility) are often linked to copy changes
This is especially true of numerous types of
cancers, where local amplifications and
translocations increase the copy number of cell
proliferation regulators, etc.
18
Amplifications in breast cancer lines increase
the copy of specific regulators ..
19
Once genome polymorphisms (SNPs, copy number,
rearrangements) are identified, one can associate
them with variation in phenotype
through association studies.
20
Genome-wide QTL mapping linking gene expression
to genoptype
21
Map the differences in phenotype between
laboratory yeast strain (BY) and a wild isolate
from a vineyard (RM).
BY
RM
Gene expression in 6 replicates BY vs. 6
replicates of RM. 1500 genes show differential
expression with p lt 0.005 (25 expected by chance)
22
Map the differences in phenotype between
laboratory yeast strain (BY) and a wild isolate
from a vineyard (RM).
BY
RM
X
Meiosis homologous recombination
Hybrid diploid
The yeast meiotic products (spores) are joined
together in a tetrad
23
Next, use DNA microarrays to identify regions of
tetrad genomes inherited from BY parent or RM
parent (IBD Identity By Descent)
By mapping IBD regions, can identify sites of
homologous recombination (crossovers)
24
Next, use DNA microarrays to identify regions of
tetrad genomes inherited from BY parent or RM
parent (IBD Identity By Descent)
Measured gene expression and IBD in 40 haploid
spores resulting from BY x RM cross.
25
Did QTL mapping to link the genotype responsible
for the gene-expression phenotype
Identified cis and trans effects Mutations in
cis those immediately at the affected
locus eg) mutations in transcription factor
binding sites Mutations in trans those
mutations that act from a distance on gene
expression eg) mutation in a transcription
factor coding sequence mutation that
causes secondary effect on gene expression.
To test these predictions measure
allele-specific expression in hybrid
strains (Ronald et al. 2005b) 60-70 of all
genes with self-linkage have mutations in cis
(and fall both upstream and downstream of
coding regions) many of these mutations are
not in known TF binding sites.
26
(No Transcript)
27
(No Transcript)
28
(No Transcript)
Recommended
CrystalGraphics Presentations





























