
Distance Based Methods for estimating phylogenetic trees - PowerPoint PPT Presentation
1 / 49
Title:
Distance Based Methods for estimating phylogenetic trees
Description:
The Jukes-Cantor model states that all states {A,C,G,T} and all ... Under the Jukes-Cantor model where all point mutations are equally likely the correction is: ... – PowerPoint PPT presentation
Number of Views:334
Avg rating:3.0/5.0
Title: Distance Based Methods for estimating phylogenetic trees
1
Distance Based Methodsfor estimating
phylogenetic trees
Phylogenetics Workhop, 16-18 August 2006
Barbara Holland
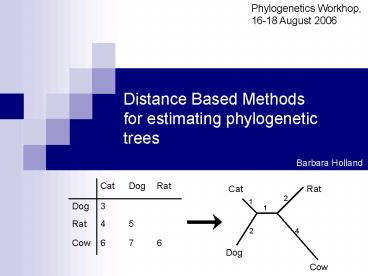
Cat
Rat
2
1
1
2
4
Dog
Cow
2
Overview
- How do we get distance data?
- Observed vs. actual distances
- Correcting for hidden changes
- Not all distances are tree-like
- Tree building clustering methods
- UPGMA
- Neighbor-joining
- Tree building optimality criteria
- Least Squares
3
What do edge lengths represent?
- In some trees edges represent time, in which case
all modern sequences should be the same distance
from the root. - Sometimes edge lengths represent the product µt
of the rate of change µ and time t in which case
different tips can be different distances from
the root provided that the rate has changed
across the tree.
Cat
Rat
2
1
1
2
4
Dog
Cow
4
Distance matrices
- There are many ways of building phylogenetic
trees, one family of methods uses a distance
matrix as a starting point. - A distance matrix is a table that indicates
pairwise dissimilarity, for instance...
5
Properties of distances
- d(x,x) 0
- d(x,y) d(y,x)
- d(x,y) d(y,z) gt d(x,z) (the triangle
inequality) - The distances used in phylogenetics always have
the first two properties but sometimes not the
third.
6
I want to build a tree - will any old distances
do?
- Not all distances will be suitable for building
trees. - Tree-building methods do not discriminate, they
will return a tree regardless of whether you give
them roadmap distances or distances based on a
sequence alignment. - Some distances are perfectly tree-like.
7
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
8
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
9
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
10
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
11
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
12
Perfectly tree-like distances
Cat
Rat
2
1
1
2
4
Dog
Cow
13
The 4-Point Condition
- Distances that fit exactly on a tree can be
characterised by a condition on any quartet i, j,
k, l (i.e. it must hold true for any 4 taxa). - We write d(x,y) for the distance between x and y.
- Given 4 taxa i, j, k, l, of the 3 sums
- d(i,j) d(k,l)
- d(i,k) d(j,l)
- d(i,l) d(j,k)
- The largest two are equal.
- Distances with this property are called additive,
because the weights on the paths along the tree
add up to the values in the distance matrix.
14
Why is this true of tree-like distances?
i
k
i
k
i
k
j
j
j
l
l
l
d(i,k)d(j,l)
d(i,j)d(k,l)
d(i,l)d(j,k)
lt
15
Clock-like distances
- An even stricter condition on distances is that
they fit on a clock-like tree. - Distances with this property are called
ultrametric.
time
d(i,k) d(j,k) gt d(i,j)
i
j
k
16
Where do we get distances from?
- Distances can be derived from Multiple Sequence
Alignments (MSAs). - The most basic distance is just a count of the
number of sites which differ between two
sequences divided by the sequence length. These
are sometimes known as p-distances.
17
Other sources of distances
- Immunological data
- Similarity between proteins A and B can be
assessed by how well the immune system responds
to B after already having seen A. - DNA/DNA hybridization
- more similar DNA hybrids "melt" at higher
temperatures - Fragment length polymorphism
- Chop DNA up using restriction enzymes.
- Amplify some fragments usign PCR
- Run the fragments out on an electrophoretic gel
- Compare profiles of different genomes
- BLAST scores
18
Observed distances usually underestimate the true
number of changes
ATTTGCGATA
Actual Changes 2 Observed Changes 2
ATTTGCGGTA
ATCTGCGATA
19
- Parallel changes
- Reversals
- Superimposed changes
ATTCGCGATA
Actual Changes 4 Observed Changes 2
ATTTGCGGTA
ATCTGCGATA
20
- Parallel changes
- Reversals
- Superimposed changes
ATTTGCGATA
Actual Changes 4 Observed Changes 2
ATTCGCGATA
ATTTGCGGTA
ATCTGCGATA
21
- Parallel changes
- Reversals
- Superimposed changes
ATTTGCGATA
Actual Changes 3 Observed Changes 2
ATTTGCGTTA
ATTTGCGGTA
ATCTGCGATA
22
Correcting for hidden changes
- Given a statistical model of how point mutations
occur it is possible to estimate the true genetic
distance from the observed distance.
23
Correcting under a simple model
- The Jukes-Cantor model states that all states
A,C,G,T and all changes between states, e.g.
A?C, are equally likely.
u/3
As a mathematical conviencence imagine we have a
rate 4u/3 of change to a random state, this
includes the possibility of a state changing to
itself.
u/3
u/3
u/3
u/3
u/3
24
A Poisson process
- The probability of no change at a site over time
t is e-4/3ut - The probability of at least one event is then 1-
e-4/3ut - The probability of at least one event that leads
to a different state from the one we started at
is ¾(1- e-4/3ut) as one time out of four we will
mutate to the same base we started with. - The expected observed distance d given a true
genetic distance of ut is d ¾(1- e-4/3ut) - Inverting this formula gives our correction D
ut -3/4 ln (1-4/3d)
25
Correcting for hidden changes
- Correction for hidden changes has been shown
(both theoretically and by simulation studies) to
improve accuracy. - However, this is not universally true.
- If data is clock-like then corrections will not
change the relative size of the distances - However, the more complicated the model is the
larger the variance (error) of the distances will
become.
26
Under the Jukes-Cantor model where all point
mutations are equally likely the correction is
Dactual ¾ ln(1 4/3dobserved)
27
(No Transcript)
28
An interesting observation
- Uncorrected distances always obey the triangle
inequality d(x,y) d(y,z) gt d(x,z) - But corrected distance do not.
- E.g. if sequences a and b differ at 10 / 100
sites and sequences b and c differ at a different
10 / 100 sites the uncorrected distances are
d(a,b) d(b,c) 0.1, d(a,c) 0.2 and the
corrected distances (under the JC model) are
D(a,b) D(b,c) 0.107, D(a,c) 0.233
29
Tree building - UPGMA
- UPGMA works by progressively clustering the most
similar taxa until all the taxa form a rooted
clock-like tree. - Find the smallest entry in the distance matrix,
say d(x,y). - Form a new internal node, z, that is a parent to
x and y and set the edge lengths from z to x and
z to y to half d(x,y). - Update the distance matrix by setting the
distances from the new node z to all the other
taxa to be the average distance between groups x
and y. - REPEAT until all groups have been joined.
30
What precisely is meant by the average distance?
- If we a joining two groups i and j that already
have ni and nj members we update the distances
using
31
Step 1 Find the smallest entry in the distance
matrix
d(i,j)
A
B
C
D
E
F
A
-
B
2
-
C
4
4
-
D
4
4
2
-
E
7
7
7
7
-
F
5
5
5
5
6
-
G
8
8
8
8
9
5
Step 2 - Cluster taxa A and B, form a new
internal node I Calculate the lengths of the new
edges d(A,I)d(B,I)1/2 d(A,B)1
B
A
A
Step 3 Update the distance matrix d(C,I)
½(d(A,C) d(B,C)) 4 etc...
1
1
B
G
I
C
D
C
F
E
D
F
E
G
32
Step 1 Find the smallest entry in the distance
matrix
d(i,j)
I (AB)
C
D
E
F
I (AB)
-
C
4
-
D
4
2
-
E
7
7
7
-
F
5
5
5
6
-
G
8
8
8
9
5
Step 2 - Cluster taxa C and D, form a new
internal node II Calculate the lengths of the new
edges d(C,II)d(D,II)1/2 d(C,D)1
A
B
B
D
A
C
Step 3 Update the distance matrix d(I,II)1/2(d(
I,C)d(I,D)) 4 d(E,II) ½(d(E,C)
d(E,D)) 7 etc...
1
1
1
1
1
1
C
I
I
II
D
E
E
F
F
G
G
33
And so on...
A
A
B
B
C
A
B
D
A
C
D
B
G
C
D
I
I
I
II
II
C
F
III
E
E
D
E
E
F
F
F
G
G
G
F
B
C
A
D
E
G
F
B
C
F
A
B
C
D
A
D
E
1
1
1
1
II
I
2.5
II
1
II
1
I
I
3.4
III
3.8
0.5
0.9
III
III
IV
V
IV
IV
V
E
0.4
VI
G
G
...until we have a rooted tree. But, is it the
right tree?
34
UPGMA is not consistent for additive distances
d(i,j)
A
B
C
D
E
F
The tree that matches the distances is not
recovered by UPGMA.
A
-
B
2
-
C
4
4
-
D
4
4
2
-
E
7
7
7
7
-
F
5
5
5
5
6
-
G
8
8
8
8
9
5
35
Inconsistency
- When a method is given perfect data but still
gets the wrong tree it is said to be
inconsistent. - UPGMA is inconsistent for data that isnt
ultrametric (clock-like). - Next well look at a method that is consistent
for any additive data.
36
Neighbor-joining (NJ)
- NJ works by progressively clustering taxa until
all the taxa form an unrooted tree. - Rather than using the distance matrix directly to
determine which taxa should be clustered at each
stage, NJ uses the S matrix where - S(i,j) (N-2)d(i,j) - R(i) - R(j)
- N is the number of taxa.
- R(i) is the sum of the ith row in the distance
matrix. - R(j) is the sum of the jth row in the distance
matrix. - Find the smallest entry in the S matrix, say
S(x,y).
37
- Form a new internal node, z, that is a parent to
x and y and calculate the edge lengths from z to
x and z to y. - d(x,z) 1/(2(N-2))(N-2)d(x,y) R(x) R(y)
- d(y,z) d(x,y) d(x,z)
- Update the distance matrix
- d(w,z) ½ (d(x,w) d(y,w) d(x,y))
- REPEAT until only two things are left to be
joined.
38
NJ Example
D
S
Step 1
R(cat) 13 R(dog) 15 R(rat) 15 R(cow) 19
e.g. S(cat,dog) (4-2)x3 13 15
-22 S(cat,rat) (4-2)x4 13 15 -20
39
NJ Example
D
S
Step 2
Step 1
Cat
Rat
Step 3 d(cat,z) ¼2d(cat,dog) R(cat)
R(dog) ¼ 6 13 15 1 d(dog,z) 3-1
2
z
Dog
Cow
40
Step 4 d(z,rat) ½ d(cat,rat) d(dog,rat)
d(cat,dog) ½ 4 5 3 3 d(z,cow) ½
6 7 3 5
Cat
Rat
z
Dog
Cow
41
Global vs Local methods
- UPGMA and NJ are local construction methods. At
each step they pick they best pair of taxa to
cluster, once a decision is made it cannot be
unmade. This makes these methods very fast. - There are also global methods for making trees
based on distances. These evaluate an optimality
criterion on each possible tree and then pick the
tree with the best score. Examples of global
methods for distance data include least squares
and minimum evolution. Because the number of
trees grows very quickly with the number of taxa,
these methods are slow.
42
Least Squares
- We would like the path lengths on the tree we
choose to be as close as possible to the
corresponding values in the distance matrix. - With additive data we can always find a tree
where the path length distances and the distance
matrix match exactly. However, most data isnt
perfect... - We can try and minimise the discrepency between
the observed distances and the tree distances
using a least squares approach.
43
A family of least squares methods
wij 1 unweighted least squares
(Cavalli-Sforza and Edwards 1967) wij
1/Dij wij 1/Dij2 (Fitch and Margoliash 1967)
44
Picking the best weights for a given tree
- The tree distances dij can be represented by the
equation
where xij,k is an indicator variable that is 1 if
edge k lies on the path from i to j and 0
otherwise. We want to find edge weights ek that
minimise
45
The indicator variables can be expressed in
matrix format
1 1 1 0 0 0 0 1 1 0 1 1 0 0 1 1 0 1 0 0 1 1 0 0 0
0 1 0 0 0 1 1 1 0 0 0 0 1 1 0 0 1 0 1 1 0 0 1 0 0
0 0 0 1 0 1 0 1 0 1 1 1 0 0 0 0 0 0 1 1
DAB DAC DAD DAE DBC DBD DBE DCD DCE DDE
e1 e2 e3 e4 e5 e6 e7
B
A
C
e1
e3
e5
e2
e4
D
e
X
e6
e7
D
E
Each row of X corresponds to a path in the
tree We can write D Xe
46
Experience the joy of linear algebra
- DXe
- XTD (XTX)e
- e (XTX)-1XTD
This assumes that the weights wij 1
47
Minimum evolution
- Uses the least squares method to fit the branch
lengths for each tree - BUT uses a different optimality criterion than
least squares. - Prefers the tree with the shortest sum of branch
lengths
48
Review
- Observed distances derived from sequence
alignments will always underestimate the true
number of mutations. Hence it is ususally a good
idea to correct for these hidden changes. - Clustering methods like UPGMA and
Neighbor-joining are very fast as they only make
local decisions and never backtrack. These
methods are often used as a starting point for
heuristic searches. - There are also optimality criteria that use
distances as input, e.g. Least squares and
minimum evolution.
49
Review
- Not all distances can be fit perfectly onto a
tree. - Methods can be inconsistent, for example for some
non-clocklike distances UPGMA is guaranteed to
recover the wrong tree. - UPGMA is consistent for clock-like distances and
NJ is consistant for any additive distances.
Recommended
CrystalGraphics Presentations





























