
Protein Structure Prediction - PowerPoint PPT Presentation
1 / 39
Title:
Protein Structure Prediction
Description:
Protein Structure Prediction Ram Samudrala University of Washington Historical perspective on comparative modelling BC excellent ~ 80% 1.0 2.0 alignment side ... – PowerPoint PPT presentation
Number of Views:49
Avg rating:3.0/5.0
Title: Protein Structure Prediction
1
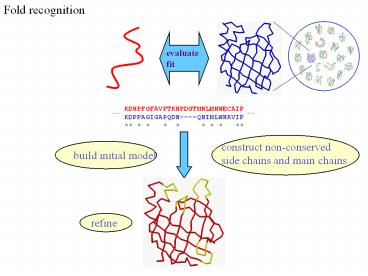
Fold recognition
refine
2
Ab initio prediction of protein structure
concept
- Go from sequence to structure by sampling the
conformational space in a reasonable - manner and select a native-like conformation
using a good discrimination function - Problems conformational space is astronomical,
and it is hard to design functions that - are not fooled by non-native conformations (or
decoys)
3
Ab initio prediction of protein structure
sample conformational space such that native-like
conformations are found
hard to design functions that are not fooled by
non-native conformations (decoys)
astronomically large number of conformations 5
states/100 residues 5100 1070
4
Sampling conformational space continuous
approaches
- Most work in the field
- Molecular dynamics
- Continuous energy minimisation (follow a valley)
- Monte Carlo simulation
- Genetic Algorithms
- Like real polypeptide folding process
- Cannot be sure if native-like conformations are
sampled
5
Molecular dynamics
- Force -dU/dx (slope of potential U)
acceleration, m a(t) force - All atoms are moving so forces between atoms are
complicated functions of time - Analytical solution for x(t) and v(t) is
impossible numerical solution is trivial - Atoms move for very short times of 10-15 seconds
or 0.001 picoseconds (ps) - x(tDt) x(t) v(t)Dt 4a(t) a(t-Dt)
Dt2/6 - v(tDt) v(t) 2a(tDt)5a(t)-a(t-Dt) Dt/6
- Ukinetic ½ S mivi(t)2 ½ n KBT
- Total energy (Upotential Ukinetic) must not
change with time
old position
old velocity
acceleration
new position
acceleration
old velocity
new velocity
n is number of coordinates (not atoms)
6
Energy minimisation
- For a given protein, the energy depends on
thousands of x,y,z Cartesian atomic - coordinates reaching a deep minimum is not
trivial - With convergence, we have an accurate
equilibrium conformation and a well-defined - energy value
7
Monte Carlo simulation
- Discrete moves in torsion or cartesian
conformational space - Evaluate energy after every move and compare to
previous energy (DE) - Accept conformation based on Boltzmann
probability - Many variations, including simulated annealing
(starting with a high temperature so - more moves are accepted initially and then
cooling) - If run for infinite time, simulation will
produce a Boltzmman distribution
8
Genetic Algorithms
- Generate an initial pool of conformations
- Perform crossover and mutation operations on
this set to generate a much larger pool of - conformations
- Select a subset of the fittest conformations
from this large pool - Repeat above two steps until convergence
9
Sampling conformational space exhaustive
approaches
enumerate all possible conformations view entire
space (perfect partition function)
must use discrete state models to minimise number
of conformations explored
computationally intractable 5 states/100
residues 5100 1070 possible conformations
10
Scoring/energy functions
- Need a way to select native-like conformations
from non-native ones - Physics-based functions electrostatics, van der
Waals, solvation, bond/angle terms - Knowledge-based scoring functions derive
information about atomic properties from a - database of experimentally determined
conformations common parametres include - pairwise atomic distances and amino acid
burial/exposure.
11
Requirements for sampling methods and scoring
functions
- Sampling methods must produce good decoy sets
that are comprehensive and include - several native-like structures
- Scoring function scores must correlate well with
RMSD of conformations (the better - the score/energy, the lower the RMSD)
12
Overview of CASP experiment
- Three categories comparative/homology
modelling, fold recognition/threading, and - ab initio prediction
- Goal is to assess structure prediction methods
in a blind and rigourous manner blind - prediction is necessary for accurate assessment
of methods - Ask modellers to build models of structures as
they are in the process of being solved - experimentally
- After prediction season is over, compare
predicted models to the experimental - structures
- Discuss what went right, what went wrong, and
why - Compare progress from CASP1 to CASP4
- Results published in special issues of Proteins
Structure, Function, Genetics 1995, - 1997, 1999, 2002
13
Comparative modelling at CASP - methods
- Alignment PSI-BLAST, FASTA, CLUSTALW - multiple
sequence alignments - carefully hand-edited using secondary
structure information - More successful side chain prediction methods
include - backbone-dependent rotamer libraries (Bower
Dunbrack) - segment matching followed by energy minimisation
(Levitt) - self-consistent mean field optimisation (Bates
et al) - graph-theory knowledge-based functions
(Samudrala et al) - More successful loop building methods include
- satisfaction of spatial restraints (Sali)
- internal coordinate mechanics energy
optimisation (Abagyan et al) - graph-theory knowledge-based functions
(Samudrala et al) - Overall model building there is no substitute
for careful hand-constructed models - (Sternberg et al, Venclovas)
14
A graph theoretic representation of protein
structure
15
Historical perspective on comparative modelling
16
Historical perspective on comparative modelling
17
Prediction for CASP4 target T128/sodm Ca RMSD of
1.0 Å for 198 residues (PID 50)
18
Prediction for CASP4 target T111/eno Ca RMSD of
1.7 Å for 430 residues (PID 51)
19
Prediction for CASP4 target T122/trpa Ca RMSD of
2.9 Å for 241 residues (PID 33)
20
Prediction for CASP4 target T125/sp18 Ca RMSD of
4.4 Å for 137 residues (PID 24)
21
Prediction for CASP4 target T112/dhso Ca RMSD of
4.9 Å for 348 residues (PID 24)
22
Prediction for CASP4 target T92/yeco Ca RMSD of
5.6 Å for 104 residues (PID 12)
23
Comparative modelling at CASP - conclusions
T128/sodm 1.0 Å (198 residues 50)
T111/eno 1.7 Å (430 residues 51)
T122/trpa 2.9 Å (241 residues 33)
T112/dhso 4.9 Å (348 residues 24)
T92/yeco 5.6 Å (104 residues 12)
T125/sp18 4.4 Å (137 residues 24)
24
Fold recognition at CASP - methods
- Visual inspection with sequence comparison
(Murzin group) - Procyon - potential of mean force based on
pairwise interactions and global dynamic - programming (Sippl group)
- Threader - potential of mean force and double
dynamic programming (Jones group) - Environmental 3D Profiles (Eisenberg group)
- NCBI Threading Program using contact potentials
and models of sequence-structure - conservation (Bryant group)
- Hidden Markov Models (Karplus group)
- Combination of threading with ab initio
approaches (Friesner group) - Environment-specific substitution tables and
structure-dependent gap penalties - (Blundell group)
25
Fold recognition at CASP - conclusions
- Fold recognition is one of the more successful
approaches at predicting structure at all - four CASPs
- At CASP2 and CASP4, one of the best methods was
simple sequence searching with - careful manual inspection (Murzin group)
- At CASP3 and CASP4, none of the threading
targets could have been recognised by the - best standard sequence comparison methods such
as PSI-BLAST - For the most difficult targets, the methods were
able to predict ? 60 residues to 6.0 Å - Ca RMSD, approaching comparative modelling
accuracies as the similarity between - proteins increased.
26
Ab initio prediction at CASP methods
- Assembly of fragments with simulated annealing
(Simons et al) - Exhaustive sampling and pruning using
knowledge-based scoring functions - (Samudrala et al)
- Constraint-based Monte Carlo optimisation
(Skolnick et al) - Thermodynamic model for secondary structure
prediction with manual docking of - secondary structure elements and minimisation
(Lomize et al) - Minimisation of a physical potential energy
function with a simplified representation - (Scheraga et al, Osguthorpe et al)
- Neural networks to predict secondary structure
(Jones, Rost)
27
Semi-exhaustive segment-based folding
EFDVILKAAGANKVAVIKAVRGATGLGLKEAKDLVESAPAALKEGVSKDD
AEALKKALEEAGAEVEVK
28
Historical perspective on ab initio prediction
29
Prediction for CASP4 target T110/rbfa Ca RMSD of
4.0 Å for 80 residues (1-80)
30
Prediction for CASP4 target T97/er29 Ca RMSD of
6.2 Å for 80 residues (18-97)
31
Prediction for CASP4 target T106/sfrp3 Ca RMSD
of 6.2 Å for 70 residues (6-75)
32
Prediction for CASP4 target T98/sp0a Ca RMSD of
6.0 Å for 60 residues (37-105)
33
Prediction for CASP4 target T126/omp Ca RMSD of
6.5 Å for 60 residues (87-146)
34
Prediction for CASP4 target T114/afp1 Ca RMSD of
6.5 Å for 45 residues (36-80)
35
Postdiction for CASP4 target T102/as48 Ca RMSD
of 5.3 Å for 70 residues (1-70)
36
Ab initio prediction at CASP - conclusions
T97/er29 6.0 Å (80 residues 18-97)
T98/sp0a 6.0 Å (60 residues 37-105)
T102/as48 5.3 Å (70 residues 1-70)
T110/rbfa 4.0 Å (80 residues 1-80)
T114/afp1 6.5 Å (45 residues 36-80)
T106/sfrp3 6.2 Å (70 residues 6-75)
37
Computational aspects of structural genomics
(Figure idea by Steve Brenner.)
38
Key points
- DNA/gene is the blueprint - proteins are the
functional representatives of genes - Protein structure can be used to understand
protein function - Large numbers of genes being sequenced - need
structures - Protein folding (from primary sequence to
tertiary structure) is a fast self-organising - process where a disordered non-functional chain
of amino acids becomes a stable, - compact, and functional molecule
- The free energy difference between the folded
and unfolded states is not very high - Experimental methods to determine protein
structures include x-ray crystallography - and NMR spectroscopy
- Theoretical methods to predict protein
structures include comparative/homology - modelling, fold recognition/threading, and ab
initio prediction - For ab initio prediction, you need a method that
samples the conformational space
39
lthttp//compbio.washington.edugt