
DNA protine - PowerPoint PPT Presentation
1 / 48
Title:
DNA protine
Description:
est la cons quence d'alt rations de l'ADN (mutation, d l tion ou remaniement ... Deux chercheurs am ricains, H. Varmus et M. Bishop, ont d couvert que les ... – PowerPoint PPT presentation
Number of Views:227
Avg rating:3.0/5.0
Title: DNA protine
1
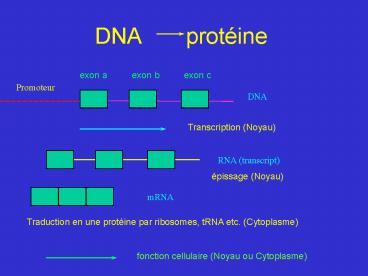
DNA protéine
2
Schéma du cycle cellulaire
3
Cancer
- est la conséquence d'altérations de lADN
(mutation, délétion ou remaniement chromosomique)
dues à - 1. Radiations
- 2. Substances chimiques
- 3. Virus (plutôt rares chez l'homme
- p. ex. Human papilloma virus)
4
Les gènes touchés sont
- 1. Ceux qui stimulent la croissance proto-oncogè
nes. - 2. Ceux qui freinent la croissance gènes
suppresseurs de tumeur. - 3. Ceux qui régulent la mort cellulaire
programmée (apoptose). - 4. Ceux qui réparent lADN.
Le cancer est un processus progressif qui
implique de nombreux gènes, donc multistep.
5
Oncogènes
- A. -Ils sont dominants et peuvent être activés
par une simple mutation sur un allèle. - B. Ils ne sont pas impliqués dans la
prédisposition héréditaire au cancer.
6
Gènes suppresseurs de tumeurs
A. Ils sont récessifs et nécessitent deux
événements pour être inactivés et donc jouer un
rôle dans la formation d'un cancer.
B. Un des deux événements peut être transmis
héréditairement. ex. Un allèle muté de p53
dans le syndrome de Li-Fraumeni ou un allèle
manquant dans le rétinoblastome
C. Réintroduction d'un allèle de type sauvage
dans une ligné cellulaire, qui ne la pas,
résulte en une ligné qui se comportent
normalement.
7
Types de Mutation
- Germinale (héréditaire).
- Mutations qui existent dans le matériel
génétique des cellules germinales, donc
transmises de père (mère) en fils (fille). - Somatique (non-héréditaire)
- Mutations qui existent dans le matériel
génétique des cellules non germinales, dont
l'effet se manifeste uniquement dans ces
cellules. Pas de transmission de père (mère) en
fils (fille).
8
Moyens d'activer un Oncogène
- Soit au niveau de la régulation de l'expression,
soit au niveau de la structure de la protéine. - 1. Mutation ponctuelle
- ex. Mutation du codon 12 du gène Ras donne
lieu à lactivation constitutive, donc sans
qu'une interaction avec un complexe
récepteur-ligand soit nécessaire.
9
Moyens d'activer un Oncogène (2)
- 2. Remaniement chromosomique soit par une
translocation soit par une inversion - A. résulte dans la surexpression de
proto- oncogène.
ex. Lymphome de Burkitt's ou le proto-oncogène
c-Myc (chromosome 8q24) passe le sous contrôle de
l'enhancer de IgH- (14q32), ceci se produit
uniquement dans les tumeurs de voie lymphocytaire
B
10
Moyens d'activer un Oncogène (3)
- B. résulte dans une protéine de fusion qui
stimule la croissance cellulaire
ex. Philadelphia translocation chromosomique
réciproque entre 9 et 22 qui génère une protéine
de fusion entre le proto-oncogène c-abl et bcr.
11
Moyens d'activer un Oncogène (4)
- 3. Amplification
- L'amplification peut aller jusqu'à une centaine
de copies du proto-oncogène. Il en résulte une
région agrandie du chromosome (HSR-homogeneous
staining region) ou dans de multiples petits
chromosomes (double minutes)
ex. N-Myc(2p) dans les neuroblastomes (30-40)
c- erb B2(egf receptor)dans les cancers du sein
(30-40) c-Myc dans les tumeurs du sein, ovaires,
et poumons cyclin D dans les carcinomes du sein
et squamous cell carcinoma
12
Découverte des Oncogènes
- Des Virus transformants
- Deux chercheurs américains, H. Varmus et M.
Bishop, ont découvert que les rétrovirus
transformants contenaient un gène supplémentaire
par rapport au même virus non transformant. - Ce gène était en réalité une copie d'un gène
cellulaire normal, un proto-oncogène, mais
modifié voir muté. C'est pour cela que les
proto-oncogènes portent souvent le nom du virus
dans lequel ils étaient trouvés, comme "fes" qui
dérive de "feline sarcoma virus". - Cette nomenclature de trois lettres pour les
oncogènes est retenue encore aujourd'hui.
13
Découverte des Oncogènes (2)
- On sait aussi que les virus peuvent transformer
les cellules par "insertion mutagenesis".
(mutagenèse par insertion) i.e. - - Soit un gène est muté par l'insertion du virus
comme dans les tumeurs mammaires induites par
MMTV (mouse mammary tumour virus). - - Soit un gène normalement silencieux peut être
activé par l'activité transcriptionelle du virus
qui s'insère adjacent à ce gène.
14
Découverte des Oncogènes (3)
- Transfection
- L'existence d'oncogènes a aussi été démontrée
expérimentalement par un autre moyen par le
chercheur américain R. Weinberg. Il a utilisé le
DNA d'une tumeur pour transformer les cellules
normales de souris. Par la suite il a pu
démontrer qu'un gène humain muté était le facteur
transformant de ces cellules. Ceci a permis la
découverte de l'oncogène ras.
15
Oncogènes et gènes suppresseurs de tumeur
16
(No Transcript)
17
Proto-oncogènes
- Ceux-ci comprennent les gènes qui régulent la
croissance - Growth Factors (Facteurs de Croissance)
- ex. Platlet Derived Growth Factor (PDGF)
- Epidermal Growth Factor (EGF)
- Growth Factor Receptors (Récepteurs de Facteurs
de Croissance) - ex. EGF récepteur
- CSF (Colony Stimulating Factor) récepteur
18
Proto-oncogènes (2)
- Protéines of signal transduction pathway
- Molécules impliquées dans la transmission d'un
signal à l'intérieur de la cellule suite à la
liaison du ligand avec son récepteur. - ex. ras, une protéine G et abl, tyrosine kinase
nucléaire - Nuclear regulatory protéines
- Activateurs de transcription ex. Myc (L, N)
- Régulateurs du cycle cellulaire
- Cyclins ex. cyclin D
- Cyclin Dependent Kinase (CDK) ex. CDK 4
19
Oncogènes et néoplasmes associés
- Oncogène Gène cellulaire Néoplasme
- Growth factors
- sis PDGF
astrocytomes, osteosarcomes - int-2 FGF sein,
melanome
Transmembrane growth factor receptors erbB
EGF receptor poumon Neu (erbB-2)
sein, ovaire, estomac fms M-CSF
leucémie
Membrane associated tyrosine kinases abl
acute myelogenous leucémie src
sarcomes
20
Oncogènes et néoplasmes associés (2)
- Membrane associated G proteines
- ras (H, K, N) carcinome du côlon, prostate,
poumons
Cytoplasmic serine-threonine kinases raf, mil
poumon
Nuclear factors c-myc Burkitts'
Lymphoma N-myc Neuroblastoma L-myc,
N-myc Small cell lung carcinome
21
Myc, Max et Mad
- Myc est un proto-oncogène souvent altéré dans les
cancers humains. - Myc peut exister comme dimère avec Max.
- Le dimère Myc-Max se lie à lADN (E-boxes) et
stimule la transcription du gène adjacent. Ceci
est applicable pour un nombre importants de gènes
comprenant CDKs, protéines ribosomales, etc. - Max-Max dimères sont inactives
22
Myc, Max et Mad (2)
- Le dimère Max-Mad, par contre, supprime la
transcription du gène adjacent. - Myc-Max favorise la croissance et Max-Mad
diminue, voire freine la croissance - Sur-expression de Myc, en labsence du signal de
proliferation, peut enclencher lapoptose. - Le taux dexpression de Myc est donc très
important pour la vie de la cellule.
23
(No Transcript)
24
Cyclines,CDKs et CDKIs
Cyclines Protéines qui sont présentes uniquement
dans certaines phases du cycle cellulaire.
Cycline A (Phase S), Cycline B (G1-M), Cycline D
(G1), Cycline E (G1)
CDK (Cyclin Dependent Kinase) Ces kinases doivent
être associés avec une cycline pour quelles
soient actives. CDK1 (cycline A B), CDK2 (A),
CDK4 (D, E), CDK6 (D,E)
CDKI (Cyclin Dependent Kinase Inhibitor),
inhibiteur sassociant avec un complexe
cycline/CDK. Il en existe 2 familles. Spectre
large p21, p27 et p57 Spectre restreint p15,
p16, p18, p19
25
Cyclins,CDKs et CDKIs Cancer
Les éléments du cycle cellulaire qui sont le plus
souvent altérés dans les cancers sont Cyclin D et
CDK4.
Cyclin D Cancer de sein, sophage (sur
expression)
CDK 4 - Mélanomes, sarcomes, et glioblastomes
(amplification)
26
Evidence pour l'existence des gènes suppresseurs
de tumeur
- 1. Des hybrides cellulaires
- A. Tumorigénique Normale Normale
B. Tumorigénique Tumorigénique
Normale Type I Type II
C. Transfert des chromosomes spécifiques dans
les lignées cellulaires tumorigéniques.
Exemple Chromosome 11 dans les lignés
cellulaires dérivées d'une tumeur de Wilm's
27
Evidence (2)
- 2. Etudes des cancers familiaux
- Des délétions d'une région chromosomique se
transmettent de façon héréditaire - Exemple13q14-Retinoblastome
3. Perte d'allèles dans les tumeurs sporadique
Délétion des même régions chromosomiques dans les
tumeurs sporadiques de même type Exemple
hémizygote pour le locus 17p13.1 (gène p53),
observé dans 70-80 des tumeurs coliques.
28
Gènes suppresseur de tumeurFonction biologique
- Facteurs de transcription
- p53 protéine (Li-Fraumeni syndrome)
- Rb, protéine du Retinoblastome
- WT 1, protéine du Wilms tumeur
2. Système de réparation de l'ADN (mismatch
repair) MLH 1 MSH2, MSH3, MSH6 PMS 1 et 2
3. Gènes régulateurs du cycle cellulaire p16
(cyclin dependant kinase inhibitor-CDKI)
29
Hypothèse de Knudson"Two Hit Hypothesis"
- 1. Deux altérations génétiques sont nécessaires
pour la formation d'une tumeur.
2. L'un des deux événements peut être transmis de
façon héréditaire.
3. Dans un cas de rétinoblastome héréditaire, le
deuxième événement se passe au niveau somatique
30
Gènes suppresseurs de tumeur
- Rétinoblastome Rb 13q14 105 kD
- Li-Fraumeni p53 17p13
53 kD - Wilm's WT-1 11p13
40 kD - Neurofibromatose NF-1 17q11 270 kD
- Polypose Familiale APC 5q21 311
kD - Melanome p16ink 9p21 16 kD
31
APC pathway
- 1. La protéine codée par le gène APC est d'une
grande taille, 310 K daltons. - 2. Il y a 10 motifs à l'intérieur de la protéine
APC qui assurent une interaction avec une autre
protéine, ß Catenine. - 3. ß Catenine, à l'état libre, peut fonctionner
comme un domaine transactivateur pour le facteur
de transcription TCF. - 4. Parmi les gènes activés par le facteur TCF on
trouve notamment cyclin D et Myc.
32
APC (2)
- Le gène APC (responsable de la maladie de
Polypose Familiale) est localisé sur le grand
bras du chromosome 5. - Ce gène est souvent muté, aussi bien dans les
tumeurs coliques sporadiques (65) que dans la
maladie de Polypose Familiale. - ß -catenine est, elle aussi, souvent trouvée
mutée dans les tumeurs coliques sporadiques
(30), les mutations sont dans la région de la
protéine qui interagit avec APC ou dans la région
qui assure une stabilité prolongée.
33
APC (3)
- Donc le taux de mutation de APC ou de ß -Catenine
est très proche de 100 dans les cancers
sporadiques du colon.
- Ceci se passe très tôt dans la progression des
tumeurs du colon
34
Rb pathway
- Tôt dans la phase G1 du cycle cellulaire, la
protéine Rb est hypophosphorylée. - Dans ce contexte, elle se lie au facteur E2F, un
facteur de transcription qui est important dans
la régulation de nombreux gènes nécessaires pour
exécuter la phase S. - Selon l'état du complexe CDK4-6 (cyclin dependent
kinase)/cyclin D, Rb est phosphorylée. - P16ink4a, un CDKi (cyclin dependent kinase
inhibitor) peut inhiber l'activité kinase du
complexe CDK4-6/cyclin D ainsi que celle de p21,
un autre CDKi.
35
Rb (2)
- Lorsque tout est en ordre dans la progression du
cycle cellulaire, Rb va être phosphorylée et
relâcher le facteur E2F pour que la cellule
puisse procéder à la phase S.
36
Rb (3)
- Il y a de nombreuses manières daltérer les voies
contrôlées par la protéine Rb
- Délétion/mutation ou suppression (par
methylation) du gène de p16
- Sur-expression de cyclin D (translocation ou
amplification)
- Mutation/délétion du gène suppresseur de tumeur
p53, donc diminution de lexpression de p21 - Mutation de CDKs.
37
Rb (4)
- Pourquoi des mutations héréditaires de Rb
donnent-elles lieu à des tumeurs de la rétine et
à des ostéosarcomes?
Labsence totale de la protéine de type sauvage
Rb enclenche l'apoptose. Mais étant donné que les
cellules de la rétine sont plus résistantes,
elles sont transformées.
38
(No Transcript)
39
p53
- Le gène p53 est le gène le plus altéré dans les
tumeurs humaines. - Le gène réside sur le petit bras du chromosome 17
(17p13) où sont observées les LOH (loss of
heterozygosity). - La protéine codée par p53 est un facteur de
transcription. - On estime que le nombre de gènes régulés par p53
est de l'ordre de 200.
40
p53 (2)
- Parmi les gènes régulés par p53 se trouvent p21
(un CDKi), BAX (une proteine accélératrice de
l'apoptose), DR5/killer (apoptose). - L'activité transcriptionnelle de p53 est
augmentée suite à différentes situations de
stress cellulaires (radiation, hypoxie, choc
thermique) suite à quoi la protéine est
stabilisée. Ceci stimule la transcription des
gènes en aval, ce qui provoque soit un arrêt du
cycle cellulaire soit l'apoptose. - Le syndrôme héréditaire, "Li-Fraumeni", est
défini par une mutation ponctuelle du gène p53.
Suite à la perte de l'allèle de type sauvage de
p53, les patients Li-Fraumeni manifestent de
nombreux types de tumeurs, notamment des tumeurs
du sein.
41
Syndromes associés aux défauts de
réparation de lADN
- Ataxia telengiectasia Le gène AT code pour une
kinase dont l activité est stimulée par des
radiations ionisantes. Parmi les protéines
activées suite à la phosphorylation par AT on
trouve p53.
- Xeroderma pigmentosum plusieurs gènes
participent à l'excision des nucléotides suite à
l'exposition aux UV.
42
Réparation du mésappariement
- HNPCC (Herediterary Non Polyposis Colon Cancer),
un exemple souvent cité, est dû aux
mutations/délétions d'un ou de plusieurs gènes
globalement appelés Mismatch repair (réparateur
de mésappariement).
- Cette classe de gènes surveille l'état de lADN
pendant la réplication. Si une erreur ce produit,
le complexe arrête la réplication et induit sa
correction.
43
Réparation du mésappariement (2)
- Les principaux gènes de la classe Mismatch
repaire sont - tumeurs sporadiques du colon
- MSH2 (2p21) 38 HNPCC.
- MLH1 (3p21) 49 (31 par methylation suppression)
- MLH3 (14q24.3) 2 (39)
- PMS1 (2q31) 0.3
- PMS2 (7p22) 2
- MSH6 (2p21) 9 (30)
44
Réparation du mésappariement (3)
- Le phénotype cancéreux se présente uniquement
dans les cellules où les deux allèles du même
gène réparateur de mésappariement sont touchés
par une mutation ou une délétion. - Leur comportement est donc, génétiquement
parlant, comme un gène suppresseur de tumeur. - La progression d'une tumeur est accélérée une
fois que les deux allèles d'un des gènes
réparateurs de mésappariement sont altérés. - L'instabilité des microsatellites est le
diagnostic moléculaire indiquant que l'un des
gènes réparateurs de mésappariement n'est plus
fonctionnel.
45
Apoptose
- Bcl-2
- Il y a un remaniement chromosomique 14q3218q21
dans 85 des lymphomes de type B. - Ce qui a pour conséquence la surexpression du
gène à 18q21 qui est bcl-2 (breakpoint cluster
2). - Ce gène est un des gènes clés de l'inhibition de
l'apoptose. Donc la surexpression due à
l'enhancer à 14q32 (IgH) protège les cellules
contre l'apoptose et permet ainsi aux cellules
avec de lADN endommagé de continuer leur cycle. - Le mécanisme par lequel bcl-2 inhibe l'apoptose
n'est pas complètement élucidé.
46
Apoptose (2)
- Bcl-2 fait partie d'une famille de gènes parmi
lesquels on trouve des gènes favorisant
l'apoptose (BAX (régulé par p53), BAD, bcl-xS) et
des gènes inhibant l'apoptose (bcl-2, bcl-xL). - L'apoptose ne dépend pas uniquement des gènes de
la famille bcl. Parmi les autres familles de
gènes, se trouve le gène DR5/killer, gène dont
l'expression dépend également de p53. - C-Myc joue aussi un rôle non négligeable dans la
décision de l'apoptose. Donc l'apoptose est un
processus assez compliqué dont il reste encore
beaucoup de détails à résoudre.
47
Génes déjà isolés dans le cancer du colon
- ras- 11p tumeurs sporadiques 1985
- p53- 17p1.3- Syndrome de LiFraumeni
- et tumeurs sporadiques 1990-92
- DCC- 18q22 tumeurs sporadiques 1990
- MCC- 5q-tumeurs sporadiques 1991
- FAP- 5q APC et tumeurs sporadiques 1991
48
Gènes déjà isolés dans le cancer du colon
- Muts- 2p21 HNPC tumeurs sporadiques 1993
- Mutl- 3p21-23 HNPC tumeurs sporadiques
1994
- PTEN- 10q22-23 Cowden's disease, tyrosine
phosphatase 1997
- SMAD 4- 18q21.1 Juvenile polyposis 1999
- LKB1- 19p13.3 Peutz-Jeghers syndrome (PJS)
, serine/threonine kinase 1999
- BMPR1A/ALK3- 10q22.3 Juvenile polyposis, TGFß
receptor family 2001
Recommended
CrystalGraphics Presentations





























